

The foremost priority of all efforts to limit the damage caused by the Coronavirus is Containment.
Public health efforts to contain the spread of the virus depend heavily on predicting how the disease is going to spread further. Since resources are finite, governments and nonprofits are attempting to deploy them in the best way to limit the spread as much as possible. Timely action in Singapore, Hong Kong and India (social distancing and travel restrictions in particular) have proven to be successful in containing the virus compared to the catastrophic effect of lax early actions in Italy, where almost 20,000 lives have been lost, and over 1.5lakh people have been affected.
Modelling the spread of COVID-19
Modern computational power allows us to build statistical models to quickly incorporate multiple inputs, such as a given disease’s ability to pass from person to person and the movement patterns of potentially infected people travelling by air and land. This process sometimes involves making assumptions about unknown factors, such as an individual’s exact travel pattern. By plugging in different possible versions of each input, however, researchers can update the models as new information becomes available and compare their results to observed patterns for the illness. For example, if investigators want to study how closing a particular airport could affect a disease’s global spread, their computers can swiftly recalculate the risk of importing cases through other airports—all the humans need to do is update the network of flight routes and international travel patterns.
Why is it difficult to model the spread of the Virus?
As is often the way with infectious diseases, modelling the spread of COVID-19 is proving to be difficult. Since the infection is relatively new, reliable data is scarce. Uncertainty about statistics such as the reproduction number (the average number of new cases caused by an infected individual) or the fatality rate can have a butterfly effect of making a model’s predictions off by several orders of magnitude.
Additionally, models now also have to take into consideration the impact of public health interventions—such as the adoption of face masks, school closures or larger governmental measures, such as China’s decision to quarantine entire cities—along with international travel bans and constraints, and also when these measures were taken—e.g a quarantine of Wuhan probably won’t be very effective now that the virus has spread to other places.

So how do these models work?
Because each unknown factor introduces more uncertainty to a model, some researchers favour focusing on a more limited model that relies on just one main factor. One such group has used international flight data (collected from official aviation databases such as India’s Directorate General of Civil Aviation) to predict which airports represent the highest-risk getaways for the virus to spread worldwide and an expected sequence of countries in which the virus would spread. Despite not factoring individual’s movements on the ground and any sort of probability factor for person-to-person transmission, the model’s predictions were very much in line with the actual spread (the model was tested after the fact).
Another recent effort to estimate how the coronavirus is spreading—both inside China and internationally—also incorporates individual mobility data from both flights and ground-travel patterns during the period of the Lunar New Year holiday (which fell on January 25 this year) when the outbreak was picking up steam. In a paper published in The Lancet on January 31, Hong Kong based researchers estimated this year’s holiday travel patterns by using information from the 2019 Lunar New Year travels of millions of people who used the WeChat app and other services owned by Chinese tech giant Tencent.
Unlike the purely travel-focused models, however, this study also included person-to-person transmission estimates, along with travel patterns based on both official flight data and Tencent’s individual mobility data. Its results suggest COVID-19 had already taken root in many major Chinese cities as of January 25 and that those cities’ international airports helped spread the virus internationally.
Another crucial aspect being studied is the impact of social distancing and nationwide lockdowns that are being imposed in many countries. One such study by Gaurav Suryanwanshi, a 4th year Undergraduate from IIT Kharagpur, uses Bayesian probability to simulate how fast the virus would spread in a city under varying levels of lockdown. The study suggests that just a 21-day lockdown might not be enough to make a serious impact on the total number of cases, and may actually disguise the true number of cases. Read a detailed analysis of how the model works in an article by Gaurav himself in this issue.
Though there are errors and pitfalls in every model, if multiple models point in the same direction, we can be reasonably sure of at least some level of correctness of the results.
Combating Fake News
Containment measures aside, it is equally important to ensure the society continues to function smoothly, to maintain Control. Amidst the COVID-19 pandemic, a lot of fake news has been surfacing, which can incite panic and in extreme cases communal unrest. Researchers are building systems to check the spread of fake news articles via social media sites. In addition to this, governments and public interest NGOs are setting up websites to provide reliable sources of information about the virus.
Researchers at IIT Bombay have launched a fact-checking platform called Kauwa-Kaate. The platform is aimed at providing an easy to use interface for an end-user to verify the validity of an article or a forwarded message. Kauwa-Kaate is accessible through multiple interfaces - users can forward articles to a WhatsApp number, or can share the articles with a mobile app, or can copy-paste the text/images into a web interface.
Given an article or image, the system crawls a number of fact-checking sites (such as AltNews, BoomLive, factly.in etc.) and reputed news outlets (Times of India, India Today, The Hindu etc.) and returns similar articles that have appeared on these sites. Based on this, the article is labelled as trustworthy.
In addition to text, the platform also accepts image or video queries. Fake news sometimes appears as a combination of text and images that may individually be valid but are taken in different contexts. Kauwa-Kaate allows the user to query the validity of the image and the text taken together. In addition to this, the system is capable of extracting text from screenshots using Optical Character Recognition (OCR) and querying the text automatically.
The team at IIT Bombay is currently working on estimating the bias and trustworthiness of the websites themselves, using references in tweets. The idea could be to evaluate an inherent bias in the site itself (particularly in political views) or bias with respect to a particular topic. They are also working on completely automating the chat-based interface.
The government has launched a WhatsApp chatbot — called MyGov Corona Helpdesk — to get instant authoritative and reliable answers to their coronavirus queries such as the symptoms of the viral disease and how they could seek help. The platform has gained an impressive 2.5 crore users as of March 30.
The bot was built by Mumbai-based firm Haptik Technologies, which local telecom giant Reliance Jio acquired last year, and the information is being provided by the Ministry of Health. Users can interact with the bot in both Hindi and English. However, the bot only accepts a few standard queries.
Haven’t such pandemics happened before?
Now that we’ve seen some symptomatic treatment efforts, we look into how we can deal with the problem of viral diseases at the root level, the Cure. The transmission of a virus from animals to humans is nothing new. A virus transmitted from an animal to a human is called a zoonotic virus, and this transmission is called a spillover event.
While the new coronavirus SARS-CoV-2 (which causes COVID-19) is the latest example, a host of infectious and deadly diseases have hopped from animals to humans and even from humans to animals. Multiple strains of H1N1 have caused pandemics, including the 2009 flu pandemic and the 1918 outbreak known as the Spanish flu. Some studies estimated that 11 to 21% of the global population at the time—or around 700 million to 1.4 billion people (of a total of 6.8 billion)—contracted the illness.

Other well-known examples are Rabies (caused mostly by dog bites, though other animals are also carriers), Ebola (believed to have originated from infected bats and non-human primates) and SARS (Severe Acute Respiratory Syndrome, transmitted to humans through civet cats), to name just a few.
This phenomenon is hardly limited to viruses; bacteria and parasites are also often transmitted in this way. The Black Death - caused by the bacterium Yersinia pestis, carried by rodents and transmitted to humans by infected fleas - is believed to have caused between 75-125 million deaths globally. On a side note, it’s interesting to note that bats have been the origin of a number of viral diseases because they are excellent hosts.
So can we use past spillover events to our advantage?
After the SARS outbreak of 2003, scientists began to pay more attention to zoonotic viruses and spillover events. This new urgency—combined with the arrival of new sequencing technologies catalyzed by the Human Genome Project—kicked off a viral discovery boom. Scientists began to sequence the genomes of viruses, particularly coronaviruses, that occur in wild animal populations around the world.
This data was made publicly available through GenBank, an annotated collection of all publicly available nucleotide sequences and their protein translations. Started in 1982, has become an invaluable tool for virologists for developing targeted treatments for diseases like Covid-19 by inhibiting the mechanisms by which the viruses infect human cells. GenBank has grown enormously in recent years, with the number of sequences doubling rough every 18 months. A quick search on the GenBank reveals more than 40,000 sequences for coronaviruses alone.
The Middle East Respiratory Syndrome (MERS), also called the Camel Flu, broke out in 2012. It was caused by a beta-coronavirus derived from bats and transmitted to humans through camels. When scientists sequenced the the MERS-coronavirus (MERS-CoV), they noticed that the protein it used to attack human cells was similar to the one used by another coronavirus called HKU4-CoV, discovered in bats in the Guangdong province of China. The genome of the HKU4-CoV had been fully sequenced in 2007.
However, no one had observed these similarities when the MERS pandemic was at its peak. If this data had been available at the time, scientists would have had a head start at figuring out how it’s transmitted and what drugs might work against it.
Coronaviruses are so named because of the array of spike proteins on their surface that, under magnification, look like a crown. Those spike proteins are what the virus uses to gain entry to host cells, where it can replicate and spread. Most coronaviruses have nearly identical spike proteins, save for the very tip of what’s called “the receptor-binding domain,” or RBD. Subtle differences in the shape of this part of the spike dictate which kinds of cells the virus can infect.
Michael Letko, a virologist at the Laboratory of Virology at the Rocky Mountain Laboratories in the U.S (Hamilton, Montana), has built a system of synthetic virus particles engineered to express a generic version of the coronavirus spike protein, in which we can change the sequences of the RBDs. These particles can enter cells like viruses but do not replicate, instead, they trigger a chemical reaction causing the cells to fluoresce yellow-green. This allows us to observe which RBDs bind to which receptor and to what extent.
Using this system of synthetic viruses, scientists can produce experimental data about whether a or not a virus can infect human cells and identify which cells the virus is most likely to infiltrate. If we can characterize the viruses that are capable of crossing the species barrier, we may be able to prevent future pandemics by isolating potential sources of infection and by developing vaccines in advance. Knowledge of similarities in the genomes of different viruses would also be instrumental in the development of vaccines.
How can we use the genome of Sars-Cov-2 virus for developing a cure?
In early January, Chinese health authorities announced they had isolated the pathogen behind the mysterious outbreak. It was a novel coronavirus, never before seen in humans. Researchers around the world pounced on the data—to try to figure out where the virus had come from and gather clues about how it was attacking human cells.
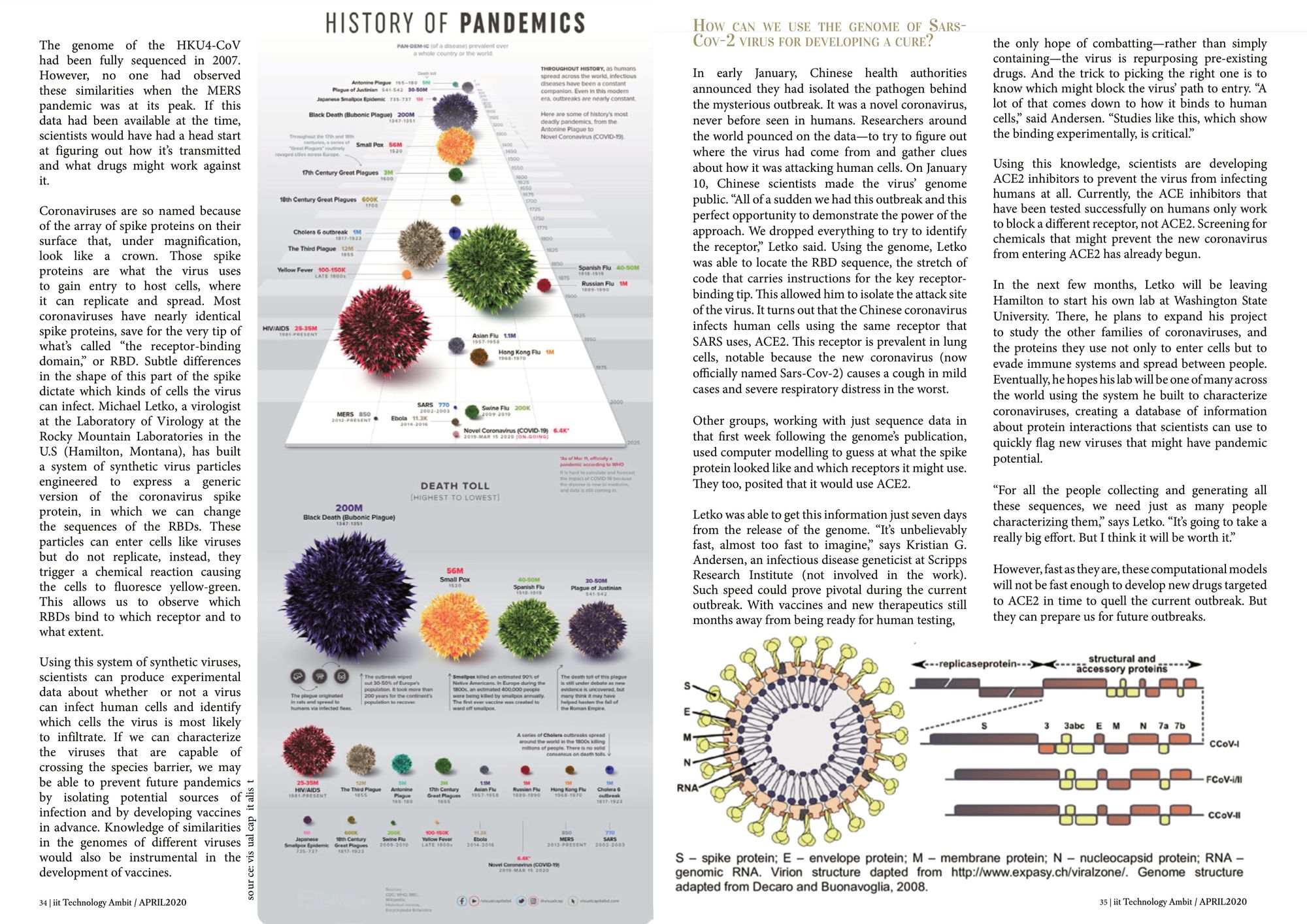
On January 10, Chinese scientists made the virus’ genome public. “All of a sudden we had this outbreak and this perfect opportunity to demonstrate the power of the approach. We dropped everything to try to identify the receptor,” Letko said. Using the genome, Letko was able to locate the RBD sequence, the stretch of code that carries instructions for the key receptor-binding tip. This allowed him to isolate the attack site of the virus. It turns out that the Chinese coronavirus infects human cells using the same receptor that SARS uses, ACE2. This receptor is prevalent in lung cells, notable because the new coronavirus (now officially named Sars-Cov-2) causes a cough in mild cases and severe respiratory distress in the worst.
Other groups, working with just sequence data in that first week following the genome’s publication, used computer modelling to guess at what the spike protein looked like and which receptors it might use. They too, posited that it would use ACE2.
Letko was able to get this information just seven days from the release of the genome. “It’s unbelievably fast, almost too fast to imagine,” says Kristian G. Andersen, an infectious disease geneticist at Scripps Research Institute (not involved in the work).
Such speed could prove pivotal during the current outbreak. With vaccines and new therapeutics still months away from being ready for human testing, the only hope of combatting—rather than simply containing—the virus is repurposing pre-existing drugs. And the trick to picking the right one is to know which might block the virus’ path to entry. “A lot of that comes down to how it binds to human cells,” said Andersen. “Studies like this, which show the binding experimentally, is critical.”
Using this knowledge, scientists are developing ACE2 inhibitors to prevent the virus from infecting humans at all. Currently, the ACE inhibitors that have been tested successfully on humans only work to block a different receptor, not ACE2. Screening for chemicals that might prevent the new coronavirus from entering ACE2 has already begun.
In the next few months, Letko will be leaving Hamilton to start his own lab at Washington State University. There, he plans to expand his project to study the other families of coronaviruses, and the proteins they use not only to enter cells but to evade immune systems and spread between people. Eventually, he hopes his lab will be one of many across the world using the system he built to characterize coronaviruses, creating a database of information about protein interactions that scientists can use to quickly flag new viruses that might have pandemic potential.
“For all the people collecting and generating all these sequences, we need just as many people characterizing them,” says Letko. “It’s going to take a really big effort. But I think it will be worth it.”
However, fast as they are, these computational models will not be fast enough to develop new drugs targeted to ACE2 in time to quell the current outbreak. But they can prepare us for future outbreaks.
