

This article is based on the discussion I had with Dr. Satyesh K. Yadav, Assistant Professor at the Department of Metallurgical and Materials Engineering, Indian Institute of Technology Madras, Tamil Nadu, India.
Background
From Nokia-1100 to iPhone 12 Pro Max, from Tata Nano to Buggatti La Voiture Noire, and from drinking glass at home to the window glass of space shuttle, we have come a long way in developing materials for a variety of applications. Stopping a Ferrari from 300 km/hr to a standstill or landing an Airbus A380 safely, materials development demands innovation across industries.
The ever increasing demand for efficient power generation and environment-friendly air travel has kept materials scientists on their toes for decades, if not for centuries. Computational Materials Science is one such field that encompasses the entire materials development cycle.
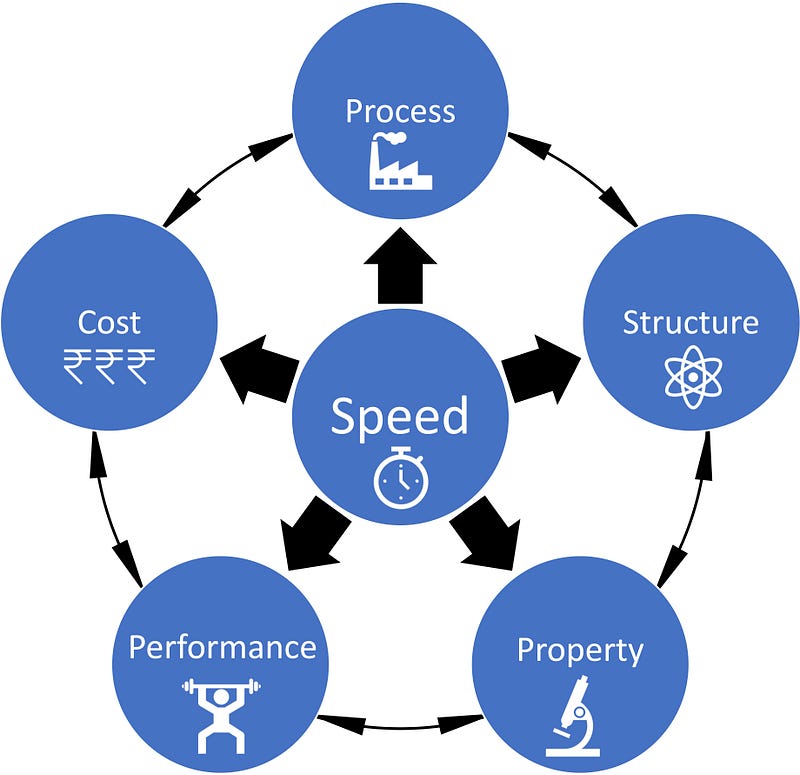
Rapid process development, quick micro-structural analysis, faster property evaluation, improved performance. All of it at a reduced cost. That's the ultimate aim of Computational Materials Science (CMS)/Integrated Computational Materials Engineering (ICME).
The revolution in computers has led to faster materials discovery and quicker alloy development cycles. Quicker and cheaper computing resources have made it possible to run an entire experiment on desktop computers instead of running them on expensive equipment.
Current State
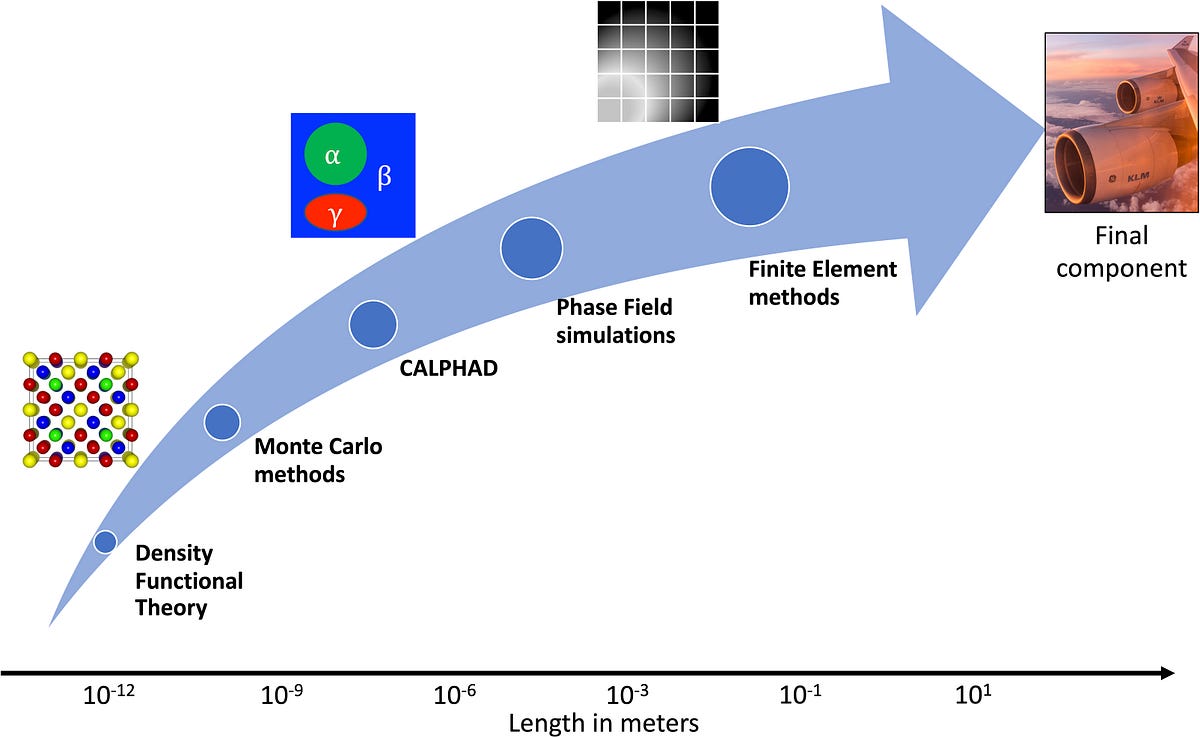
From simulating materials' electronic properties using ab-initio Density Functional Theory techniques to testing the entire component in a Universal Testing Machine, CMS/ICME covers materials development at all length scales.
Multi-scale modeling is the primary area of Computational Materials Science (CMS)/Integrated Computational Materials Engineering (ICME), where the materials are modeled/simulated/tested right from their electrons to the bulk component spanning meters. CMS/ICME techniques at various length scales are:
- Density Functional Theory (DFT) — Electrons, Atoms — Picometers
Atomistic modeling technique based on fundamental Schrödinger’s Quantum Mechanics equation. The ultimate technique to model/simulate, atoms/electrons free from any assumptions.
Requires expensive supercomputers to run the simulations. Can’t handle more than a few hundred atoms. Works at atomic length scales. - Molecular Dynamics (MD)—Atoms, Molecules — Angstrom, Nanometers
Another atomistic modeling technique based on Newton’s Laws of Motion. Requires inputs from DFT.
Can smoothly run on desktop computers. Can handle a few million atoms. Works at molecular length scales. - Calculations of Phase Diagram (CALPHAD) — Materials stability—Microns
Bulk materials modeling technique based on Laws of Thermodynamics and Kinetics. Calculates relative stability of various materials phases at equilibrium.
Can smoothly run on personal computers. Works at the micron/microstructure level. - Phase Field — Grains, Interfaces — Microns
Similar to CALPHAD, but takes into account the local interactions at different interphases as well.
Requires workstations. Works at micron/microstructure level. - Finite Element Methods — Component level — Meters
Bulk materials modeling technique based on mechanical and thermal properties of materials.
Requires workstations. Works at component level in millimeters and as well as the entire bulk structure level in meters.
Future Outlook
With the revolution in data sciences, the Computational Materials Science field also has been positively affected. Faster computers and cheaper computational resources combined with developments in Data science, Artificial-Intelligence/Machine-Learning approaches, are further reducing the time-to-market. Now we understand our components/materials in a much better way than we used to a decade ago.
Right now, it is almost impossible to model even a mole of atoms in the ab-initio Density Functional Theory; that's 10 followed by 23 zeroes. But a kilogram of iron has more than 17 moles of iron atoms. That means right now we cannot even model a gram of iron using the fastest supercomputer available in the world!
Another example is of Rolls-Royce, which generates three petabytes of data every year from its jet-engine fan blade manufacturing alone! [1] This is equivalent of a stack of CDs 3 miles high! A thorough analysis of this much data can lead to major improvement in fuel efficiencies and cost savings.
Figure below explains the alloy development through CMS/ICME approach.
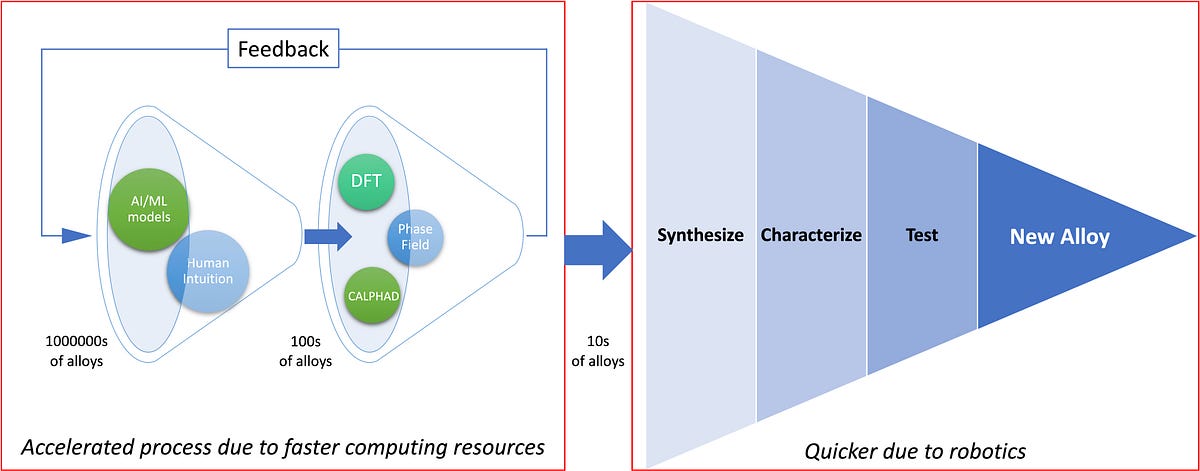
In the future (which is almost here), millions of alloy compositions will be studied using complex AI/ML models. These models filter the alloys based on experimentally reported data and then fed them to Physics and Thermodynamics based simulations, which further filters down the alloy compositions to be synthesized and tested.
We will be seeing more and more Physics being taken into account for higher and higher length scales in these computational techniques. The innovations in AI/ML techniques will lead to decreased human interference in the alloy development cycles, which act as a bottleneck right now.
We expect the entire computational materials science field to get automated in the future to the point that once the design parameters are set, the machines can do the alloy searching to synthesis to the testing, themselves. Quicker feedback from simulations-synthesis-testing stages, across the length scales will lead to faster development of new materials.
As rightly put by Dr. Satyesh K. Yadav from Indian Institute of Technology Madras:
“We don't even have a storage, forget about the processing a mole of atoms right now. By any standard Computational Materials Science (CMS) and Integrated Computational Materials Engineering (ICME) will play a crucial role in the future of materials development. As the capabilities of each of the materials modelling techniques increase over larger length and time scales, they will start overlapping more and more. The gap between atomistic modeling techniques like Density Functional Theory, and CALPHAD techniques would start fading. The fidelity of ICME would improve, and we will be able to accommodate more and more physics to larger length scales.”
— Dr. Satyesh K. Yadav
Assistant Professor
Department of Metallurgical and Materials Engineering
Indian Institute of Technology Madras, Tamil Nadu
India.
